
Lecture 3: Genetic Variation
Contents
Lecture 3: Genetic Variation¶
Molecular Genetics¶
The transmission rules of genetic material are of fundamental importance to the evolution, the theory of natural selection, and to all components of population and quantitative genetics. 90 years ago, before we knew the molecular structure of DNA, or even that DNA was the source of heritable variation, it was possible to do genetics and to properly think about the ways in which genetic variation was transmitted and modulated over evolutionary time. Ask yourself- how could this be so? These days however it is essential that you have a solid background in the basics of both molecular and mendelian genetics.
DNA is the genetic material. A phosphate sugar backbone with purine or pyrimidine bases bound to the sugar moiety. In the double helix structure of DNA, Adenine pairs with Thymine and Cytosine pairs with Guanine. A:T or C:G pairs are held together with hydrogen bonds, the C:G pairing being stronger than the A:T pairing.
The eukaryotic Chromosomes are made up of chromatin which is about half protein (histones) and half DNA. The DNA is coiled around the histones in a nucleosome structure, and these strings of nucleosomes are then coiled upon themselves making the hierarchical packaging of the genetic material very space-efficient. Recently it has come to light that nucleosome positioning on chromosomes not only “packs” DNA, but directly influences the regulation (e.g. transcription) of that DNA. The prokaryotic “chromosome” (usually one per cell) does not have the tight packaging of eukaryotic chromosomes, but is associated with various proteins.
The Central Dogma describes the general view that information transfer in genetics is unidirectional from DNA to RNA to protein, and has come to refer to the general mechanisms by which this information is retrieved. Transcription is the polymerization of a strand of RNA from DNA by the enzyme RNA polymerase. Translation is the various mechanisms by which the sequence of nucleotides in the RNA is translated into a polypeptide and requires transfer RNAs, the messenger RNA, ribosomes, amino acids (see Futuyma Figure 8.1).
The original view of the central dogma was one gene \(\rightarrow\) one protein (one gene codes for the production, through transcription and translation, of one protein). This has been modified to the view of one gene \(\rightarrow\) one polypeptide since some genes code for parts of a protein; and further defined as one gene \(\rightarrow\) one function, since some genes code for RNA products (ribosomal RNA, transfer RNA) which clearly are not polypeptides. Protein genes can be structural (e.g., collagen in your skin); catalytic (e.g., amylase enzyme in your saliva). How would you classify hemoglobin?? RNA genes can also be structural (ribosomal RNA), catalytic (RNase P acts as a cleavage enzyme).
Gene structure in prokaryotes often takes the form of an operon which is a set of adjacent structural and regulatory genes. The coding regions (= genes) are uninterrupted open reading frames of DNA that are transcribed as one RNA and translated into distinct polypeptides. The adjacent regulatory regions can alter the expression of the genes in response to specific signals from the cell.
Gene structure in eukaryotes is quite different , most notably in that the coding regions are often broken up into exons (expressed) and introns (intervening sequences). Other specific sequences in the DNA can serve as promoter elements to stimulate transcription. The intron/exon structure of eukaryotic genes means that after transcription into RNA (called the pre-messenger RNA), the intron sequences in the RNA must be removed and the exon sequence spliced back together. After splicing, the RNA is called the messenger RNA (or “mature” messenger RNA) and is transported out of the cell nucleus into the cytoplasm where it will be transcribed into protein.
The translation process goes on at the ribosomes and woks according to the rules of the genetic code. With 20 amino acids found in proteins and only 4 nucleotides (A,C,T,G), the DNA must be read in blocks of 3 nucleotides to provide enough information to translate DNA into polypeptides (\(4^1\) only = 4 ; \(4^2\) = 16 [still 4 amino acids short]; \(4^3 = 64\) [more than needed]). The very important result of the fact the \(4^3 = 64\) means the genetic code is a redundant code, i.e., some three-nucleotide “words” will translate into the same amino acid (see Futuyma Figure 8.2). This means that some nucleotide positions in the coding region of a gene will be silent with respect to the amino acid sequence that gets translated from that DNA \(\rightarrow\) RNA. “Silent” means that if a mutation occured at such a position and changed the nucleotide, the resulting amino acid sequence would still be the same (e.g., see the third position for Leu in the table). Other sites will affect the amino acid sequence if mutated (see second positions for all codons in the code). We can thus refer to two general types of nucleotide positions in the DNA of coding regions silent or synonymous sites (if mutated, no amino acid change) and replacement or nonsynonymous sites (if mutated, a different amino acid sequence will result). One consistent observation from evolutionary genomics is that synonymous sites evolve more rapidly than nonsynonymous sites in genes– why do you think this is so?
If we tabulate all possible changes in a theoretical “random” coding sequence, 61 codons could code for amino acids (3 codons of the 64 total code for stop codons) with 3 nucleotides per codon = 183 nucleotides. Each of these 183 different types of positions could change 3 ways (A could mutate to C, G or T; C could mutate to A, G or T, etc.) for a total of 549 possible types of changes in a random coding sequence. If we now look at the genetic code and ask whether each of these possible changes is a silent or a replacement site, the following is obtained: 1st position: 4% silent, 96% replacement; 2nd position: 0% silent, 100% replacement; 3rd position: 69% silent, 31% replacement. This means that mutation at 3rd positions in codons will be much less likely to affect protein sequence and hence much less likely to be subject to natural selection (more later on Molecular Evolution).
The genome is the total genetic complement of the cell. In humans this is about 3 billion nucleotides in a haploid genome (sperm or egg cell). Given a rough estimate of 30,000 genes per genome and 1000 base pairs (bp) per gene this adds up to \(3\times10^4genes \times 10^3bp = 3\times10^7\)bp in genes, or only 1% of our genomes! So what the heck is the rest of the genome for?!? This is an open question currently, but it appears at this time that much of it is repetitive DNA, and a smaller fraction of it might be regulatory in nature. We’ll get back to this when we cover Molecular evolution.
Mendelian Genetics¶
Adult human (mice, fruit flies, etc.) are diploid organisms having received one set of chromosomes from the mother and one from the father. Humans have 46 chromosomes in 23 pairs; each pair is made up of one maternal and one paternal chromosome. The sex chromosomes are a “pair” in females (two X chromosomes), but in males the X and Y sex chromosomes are very different in size. The other 22 pairs of chromosomes are autosomes.
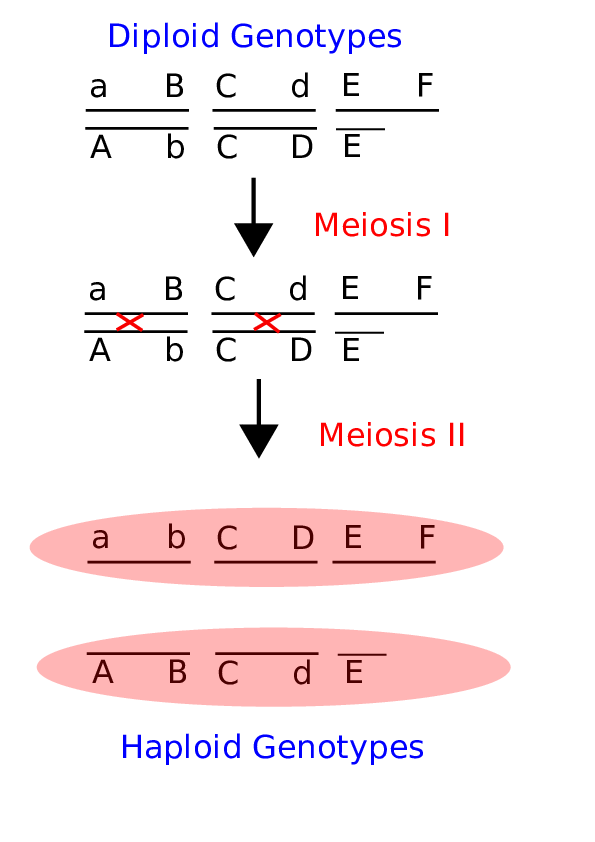
Fig. 3 Meiosis and recombination Here is a brief cartoon that illustrates the important aspects of Meiosis for the purposes of understanding how recombination (sex) shuffles alleles between chromosomes to create new genotypes. Crossover events are shown as red X’s in the middle panel.¶
Consider Figure 1 above. Each chromosome will undergo independent assortment through meiosis, thus they need not segregate as a bottom and a top pair. This alone creates diploid genotypic variation, however recombination during Meiosis (shown as red crossovers) creates new allelic combinations to create haploid genotypic variation. Why would this be important or even selected for itself? Recombination in many ways is the purpose of sex, and we’ll cover the evolutionary origins of sex later on in the class. Note that the lower gamete does not carry an F locus. This is the case in males where there is no section of the Y chromosome that is homologous to the X chromosome.
Crosses between genotypes are best diagrammed in a Punnet square. These are made by putting all possible types of gametes of one parent along one side of the square and all possible types of gametes of the other parent along the other side. The boxes are filled in by pairing gametes together to make genotypes. In a cross of two AA homozygotes, all offspring are identical. In a cross between two heterozygous Aa x Aa, each parent can produce two possible gametes: A or a. In the cross the genotypes come out as 1:2:1 of AA:Aa:aa. In a two locus situation where the loci are on separate chromosomes (unlinked) and each locus is heterozygous, each adult can produce 4 different gametes: AB, Ab, aB and ab. These four gametes go along each edge of a 4x4 Punnet square resulting in 16 possible 2-locus genotypes: AABB, AABb. AaBB, etc (figure 2.6, pg. 32). How would the Punnet square be different if the two parents had these genotypes: AABb x AaBB? Work it out!
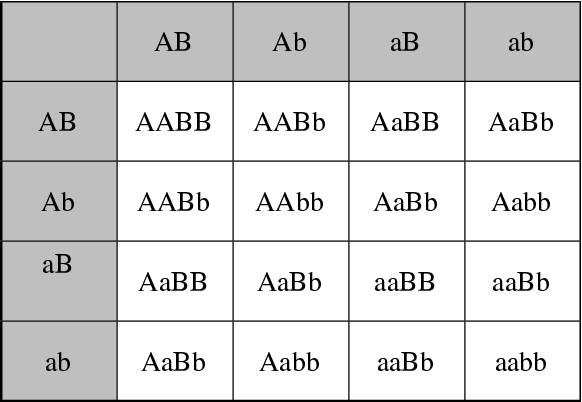
Fig. 4 A Punnet Square for a dihybrid cross This Punnet square considers the genotypes produced by a cross between two double heterozygotes, where the A and B loci are assumed to be unlinked.¶
An important issue for Darwin was the mode of transmission of traits. The Lamarkian view of the “inheritance of acquired characteristics” was workable under the Darwinian view, but was wrong. Before the rediscovery of Mendel’s work at the turn of the century, blending inheritance was accepted mainly because offspring tended to look like a mixture of parents. But note that if blending inheritance was the mechanism, then all individuals in the population would eventually come to look alike and there would be no variation upon which to select and Natural Selection would not operate. This troubled Darwin to his death. Unfortunately Darwin never found out about Mendel’s experiments even though they were published long before Darwin’s death. Mendel’s proposal of a “particulate” mechanism of inheritance solved the problem but the history of Darwinism would have been interestingly different if Darwin had understood genetics before he died. A simple example of a cross between a red (RR) and a white (rr) flower illustrates the point: RRxrr gives all Rr (lets say they are pink). Now a cross of two pink flowers: RrxRr gives back the parental types 1:2:1 RR:Rr:rr. The blending is a function of gene expression, not gene inheritance.